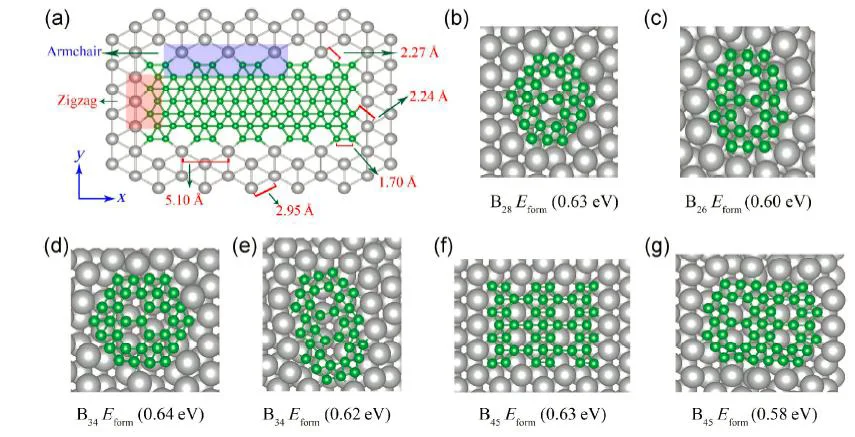
Semiconductor nanocrystals (NCs) have been greatly attractive and intensively investigated, which have brought such wide applications as nanoscale electronic and optical devices. Properties of nanomaterials mostly depend on their structures, however, it is challenging to determine the stable nanostructures from numerous possible candidates due to two main obstacles: (i) the accurate calculation of the total energy is necessary but often computationally expensive, and (ii) many isomeric structures should be considered and the number of these structures increases sharply as the size increases.
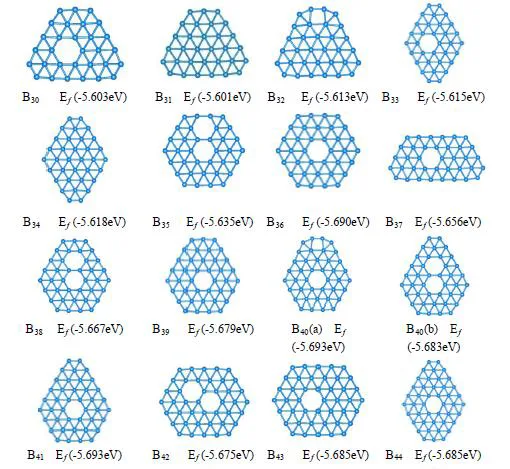
We have developed an effective model[1] to investigate the energetic stability of hydrogenated group-IV nanostructures, followed by the validations from density-functional theory calculations. The magic structures of diamond nanocrystals(CmHn) and silicon nanocrystals(SimHn) are determined, which are in agreement with the experimental observations[2,3]. There is a linear relationship of cohesive energy on the ratio (n/m) and the simplex method is used to determine the lowest energy from a set of possible integer combinations (m,n), where n<=2m + 2. The key task is to determine the lower limit of n for a certain m, which can be realized by allowing X atoms on the surface to walk randomly in the crystal lattice and saturate the configuration with H atoms where necessary, and searching the local minimum of n by iterations.
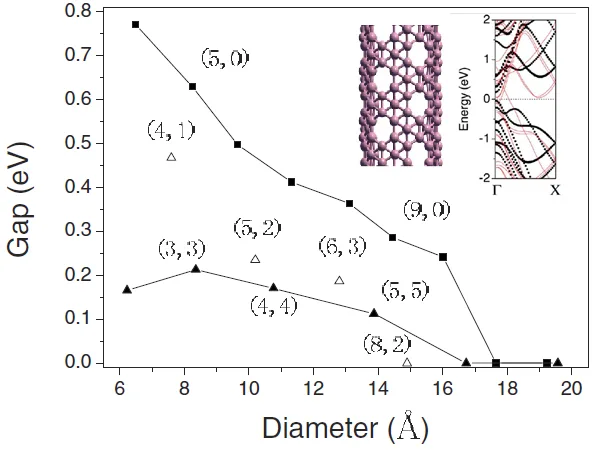
Considering the Si/Ge interactions and mixing entropy[4], the composition profiles of Si/Ge distributions in the nanowires passivated by fluorine (F)/chlorine (Cl)/hydrogen (H) atoms are obtained, which indicates the outmost layer of surface should be mostly occupied by Si. With total Si surface segregation, the diameter and shape of most stable nanowires are found to be determined by the composition x and the passivants’ chemical potential.
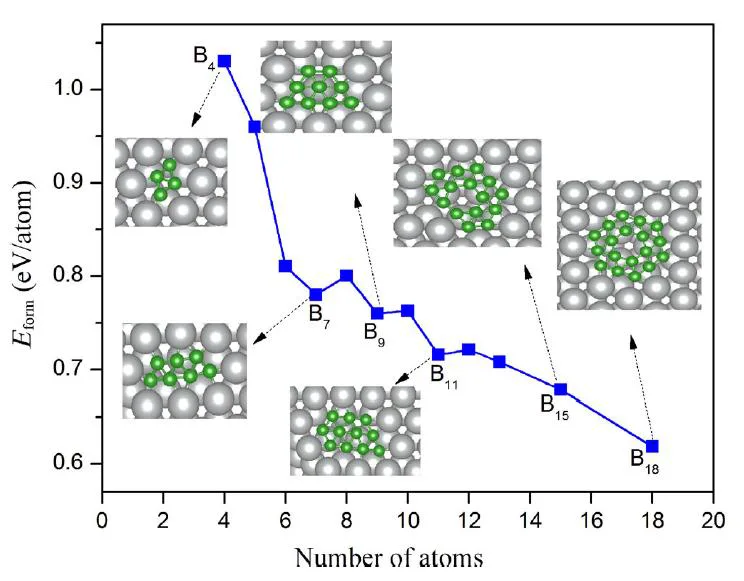
We propose an efficient approach to determine the stable H-SiCNCs by the convex analysis with the possible candidates pre-screened by the Wang–Landau method and a bond energy model[5]. We find that the configurations of H-SiCNCs are dominated by the hydrogen and carbon chemical potentials according to the phase diagram, and there are structural transitions with the increasing size from tetrahedron, hexahedron, to octahedron. Our finding indicates the possibility of designing electronic nano-devices by modulating the configurations with the control of hydrogen and carbon chemical potential
References
[1] X. Yang et. al, Phys. Rev. B, 83, 205314 (2011); 86, 085440(2012).
[2] L. Landt et. al , Phys. Rev. Lett. 103, 047402 (2009).
[3] G. Belomoin et. al, Appl. Phys. Lett. 80, 841 (2002).
[4] X. Yang et. Al J. Chem. Phys. 139, 154713 (2013).
[5] Y. Wang and X. Yang et al J. Phys. D: Appl. Phys. 49, 245305(2016).